Real Dashboards: Adding a Reset Button
Finally, let’s add a reset button to our dashboard that allows users to clear the current molecule,
header information, and histogram from the dashboard and return to the initial state. This will make
it easier for users to start fresh without having to manually clear the input field or refresh the page.
To do this, we will add a new button to our layout (to the right of the Load Structure button) and
then update our callback function to handle the reset functionality.

PDB dashboard layout with reset button.
Updated Imports
To implement the reset functionality, we will need to import the ctx object from Dash which allows
us to determine which input triggered the callback. This will help us differentiate between the load
button and the reset button. Let’s update our imports in the app.py file.
from dash import Dash, Input, Output, State, callback, ctx, dcc, html
Updated Layout
Next, we will update our layout to include a new reset button. We will use another dbc.Button component for this reset button and place it to the right of the existing load button. To line it up nicely, we will wrap both buttons in a dbc.Row component and each button in its own column using dbc.Col components.
dbc.Row([
dbc.Col(dbc.Button("Load Structure", id='load-button', color="primary"), width="auto"),
dbc.Col(dbc.Button("Reset", id='reset-button', color="danger"), width="auto"),
], className="g-2"),
Updated Callback Function
The first thing we need to do is add an additional input to our callback function for the reset button. This will allow us to trigger the same callback function when the reset button is clicked.
@callback(
[Output('molecule-viewer', 'children'),
Output('header-info', 'children'),
Output('amino-acid-histogram', 'figure'),
Output('amino-acid-histogram', 'style'),
Output('status-message', 'children')],
Input('load-button', 'n_clicks'),
Input('reset-button', 'n_clicks'),
State('pdb-input', 'value'),
prevent_initial_call=True
)
Since we now have two buttons that can trigger the same callback function, we will need to update our callback
function, load_molecule, to determine which button was clicked and handle the logic accordingly. We will use
the ctx object to check which input triggered the callback. If the reset button was clicked, we will return the
initial state of the dashboard with empty molecule viewer, header information, and histogram. If the load button
was clicked, we will proceed with loading the molecule as before.
def load_molecule(load_clicks, reset_clicks, pdb_id):
if not pdb_id:
return (
html.Div("Please enter a valid PDB ID.", className="text-center text-muted mt-5"),
html.Div("Header information will appear here.", className="text-center text-muted mt-5"),
{},
{'display': 'none'},
dbc.Alert("Please enter a PDB ID.", color="warning")
)
if ctx.triggered_id == 'reset-button':
return (
html.Div("Enter a PDB ID and click 'Load Structure' to view the molecule.", className="text-center text-muted mt-5"),
html.Div("Header information will appear here.", className="text-center text-muted mt-5"),
{},
{'display': 'none'},
None
)
Running the Updated App
Once again, putting all of these updates together, our updated app.py file should look like this:
Code
1import os
2from collections import Counter
3
4import dash_bio as dashbio
5import dash_bootstrap_components as dbc
6import plotly.express as px
7from Bio.PDB import PDBList, PDBParser, parse_pdb_header
8from dash import Dash, Input, Output, State, callback, ctx, dcc, html
9from dash_bio.utils import PdbParser as DashPdbParser
10from dash_bio.utils import create_mol3d_style
11
12# Initialize the Dash app
13external_stylesheets = [dbc.themes.CERULEAN]
14app = Dash(__name__, external_stylesheets=external_stylesheets)
15
16# App layout
17app.layout = dbc.Container([
18 dbc.Row([
19 html.Div("Molecular Structure Viewer", className="text-primary text-center fs-3 mb-4")
20 ]),
21
22 dbc.Row([
23 dbc.Col([
24 dbc.Label("Enter PDB ID:", className="fw-bold"),
25 dbc.Input(
26 id='pdb-input',
27 type='text',
28 placeholder='e.g., 4HHB, 3AID, 2MRU, 4K8X',
29 value='4HHB',
30 className="mb-2"
31 ),
32 dbc.Row([
33 dbc.Col(dbc.Button("Load Structure", id='load-button', color="primary"), width="auto"),
34 dbc.Col(dbc.Button("Reset", id='reset-button', color="danger"), width="auto"),
35 ], className="g-2"),
36 html.Div(id='status-message', className="mt-3")
37 ], width=2),
38
39 dbc.Col([
40 html.Div(id='molecule-viewer', children=[
41 html.Div("Enter a PDB ID and click 'Load Structure' to view the molecule.",
42 className="text-center text-muted mt-5")
43 ])
44 ], width=5),
45
46 dbc.Col([
47 html.Div(id='header-info', children=[
48 html.Div("Header information will appear here.",
49 className="text-center text-muted mt-5")
50 ], style={'maxHeight': '600px', 'overflowY': 'auto'})
51 ], width=5)
52 ], className="mt-4"),
53
54 dbc.Row([
55 dbc.Col([
56 dcc.Graph(id='amino-acid-histogram', figure={}, style={'display': 'none'})
57 ], width=12)
58 ], className="mt-4"),
59], fluid=True)
60
61# Callback to load and display molecule
62@callback(
63 [Output('molecule-viewer', 'children'),
64 Output('header-info', 'children'),
65 Output('amino-acid-histogram', 'figure'),
66 Output('amino-acid-histogram', 'style'),
67 Output('status-message', 'children')],
68 Input('load-button', 'n_clicks'),
69 Input('reset-button', 'n_clicks'),
70 State('pdb-input', 'value'),
71 prevent_initial_call=True
72)
73def load_molecule(load_clicks, reset_clicks, pdb_id):
74
75 if not pdb_id:
76 return (
77 html.Div("Please enter a valid PDB ID.", className="text-center text-muted mt-5"),
78 html.Div("Header information will appear here.", className="text-center text-muted mt-5"),
79 {},
80 {'display': 'none'},
81 dbc.Alert("Please enter a PDB ID.", color="warning")
82 )
83
84 if ctx.triggered_id == 'reset-button':
85 return (
86 html.Div("Enter a PDB ID and click 'Load Structure' to view the molecule.", className="text-center text-muted mt-5"),
87 html.Div("Header information will appear here.", className="text-center text-muted mt-5"),
88 {},
89 {'display': 'none'},
90 None
91 )
92
93 try:
94 # Clean up PDB ID (remove whitespace, convert to lowercase)
95 pdb_id = pdb_id.strip().lower()
96
97 # Create PDB directory if it doesn't exist
98 pdb_dir = './pdb_files'
99 os.makedirs(pdb_dir, exist_ok=True)
100
101 # Download PDB file using BioPython
102 pdbl = PDBList()
103 pdb_file = pdbl.retrieve_pdb_file(pdb_id, pdir=pdb_dir, file_format='pdb')
104
105 # Read PDB file content for visualization
106 dash_parser = DashPdbParser(pdb_file)
107 pdb_data = dash_parser.mol3d_data() # Get data in format suitable for Molecule3dViewer
108 # create styles for visualization needed by Molecule3dViewer
109 # atoms is a list of dictionaries obtained from parsing the PDB file with DashPdbParser
110 # visualization_type can be 'cartoon', 'stick', 'sphere'
111 # color_element can be 'residue', 'chain', 'element', 'partialCharge'
112 styles = create_mol3d_style(
113 pdb_data['atoms'], visualization_type='cartoon', color_element='residue'
114 )
115
116 # Parse PDB structure for amino acid analysis
117 bio_parser = PDBParser(QUIET=True)
118 structure = bio_parser.get_structure(pdb_id, pdb_file)
119 amino_acid_counts = count_amino_acids(structure)
120
121 # Parse PDB header information
122 header_info = parse_pdb_header(pdb_file)
123
124 # Create Molecule3dViewer component
125 viewer = create_molecule_viewer(pdb_data, styles)
126
127 # Create header display
128 header_display = create_header_display(header_info, pdb_id)
129
130 # Create amino acid histogram
131 histogram = create_amino_acid_histogram(amino_acid_counts, pdb_id)
132
133 status = dbc.Alert(
134 f"Successfully loaded PDB ID: {pdb_id.upper()}",
135 color="success"
136 )
137
138 if not histogram:
139 return viewer, header_display, histogram, {'display': 'none'}, status
140 else:
141 return viewer, header_display, histogram, {'display': 'block'}, status
142
143 except Exception as e:
144 error_msg = dbc.Alert(
145 f"Error loading PDB {pdb_id.upper()}: {str(e)}",
146 color="danger"
147 )
148 empty_viewer = html.Div(
149 "Failed to load molecule. Please check the PDB ID and try again.",
150 className="text-center text-muted mt-5"
151 )
152 empty_header = html.Div(
153 "Header information will appear here.",
154 className="text-center text-muted mt-5"
155 )
156 return empty_viewer, empty_header, {}, {'display': 'none'}, error_msg
157
158def create_molecule_viewer(pdb_data, styles):
159 """Create a Molecule3dViewer from PDB data"""
160 return dashbio.Molecule3dViewer(
161 id='molecule-3d',
162 modelData=pdb_data,
163 styles=styles,
164 selectionType='atom',
165 backgroundColor='#F0F0F0',
166 height=600,
167 width='100%'
168 )
169
170def create_header_display(header_info, pdb_id):
171 """Create a formatted display of PDB header information"""
172 header_sections = []
173
174 # Title
175 if 'name' in header_info:
176 header_sections.append(
177 html.Div([
178 html.H6("Name", className="fw-bold mt-3 mb-2"),
179 html.P(header_info['name'], className="text-sm")
180 ])
181 )
182
183 # Structure Classification
184 if 'structure_method' in header_info:
185 header_sections.append(
186 html.Div([
187 html.H6("Method", className="fw-bold mt-3 mb-2"),
188 html.P(header_info['structure_method'], className="text-sm")
189 ])
190 )
191
192 # Release Date
193 if 'release_date' in header_info:
194 header_sections.append(
195 html.Div([
196 html.H6("Release Date", className="fw-bold mt-3 mb-2"),
197 html.P(header_info['release_date'], className="text-sm")
198 ])
199 )
200
201 # Deposition Date
202 if 'deposition_date' in header_info:
203 header_sections.append(
204 html.Div([
205 html.H6("Deposition Date", className="fw-bold mt-3 mb-2"),
206 html.P(header_info['deposition_date'], className="text-sm")
207 ])
208 )
209
210 # Resolution
211 if 'resolution' in header_info and header_info['resolution'] is not None:
212 header_sections.append(
213 html.Div([
214 html.H6("Resolution (Å)", className="fw-bold mt-3 mb-2"),
215 html.P(f"{header_info['resolution']:.2f}", className="text-sm")
216 ])
217 )
218
219 if 'journal_reference' in header_info and header_info['journal_reference']:
220 journal_text = header_info['journal_reference']
221 header_sections.append(
222 html.Div([
223 html.H6("Journal Reference", className="fw-bold mt-3 mb-2"),
224 html.P(journal_text, className="text-sm", style={'wordWrap': 'break-word'})
225 ])
226 )
227
228 # Keywords
229 if 'keywords' in header_info and header_info['keywords']:
230 keywords_text = header_info['keywords']
231 header_sections.append(
232 html.Div([
233 html.H6("Keywords", className="fw-bold mt-3 mb-2"),
234 html.P(keywords_text, className="text-sm", style={'wordWrap': 'break-word'})
235 ])
236 )
237
238 if header_sections:
239 return dbc.Card([
240 dbc.CardBody([
241 html.H5(f"PDB: {pdb_id.upper()}", className="card-title"),
242 html.Hr(),
243 *header_sections
244 ])
245 ], style={'height': '100%'})
246 else:
247 return html.Div("No header information available.", className="text-center text-muted mt-5")
248
249def count_amino_acids(structure):
250 """Count amino acid frequencies in a PDB structure"""
251 # Standard amino acids (3-letter codes)
252 standard_aa = {
253 'ALA', 'CYS', 'ASP', 'GLU', 'PHE', 'GLY', 'HIS', 'ILE', 'LYS', 'LEU',
254 'MET', 'ASN', 'PRO', 'GLN', 'ARG', 'SER', 'THR', 'VAL', 'TRP', 'TYR'
255 }
256
257 amino_acids = []
258
259 # Iterate through all residues in all chains
260 for model in structure:
261 for chain in model:
262 for residue in chain:
263 # Get residue name and check if it's a standard amino acid
264 res_name = residue.get_resname().strip()
265 if res_name in standard_aa:
266 amino_acids.append(res_name)
267
268 # Count frequencies
269 return Counter(amino_acids)
270
271def create_amino_acid_histogram(amino_acid_counts, pdb_id):
272 """Create a Plotly histogram of amino acid frequencies"""
273 if not amino_acid_counts:
274 return {}
275
276 # Convert to lists for plotting
277 amino_acids = list(amino_acid_counts.keys())
278 counts = list(amino_acid_counts.values())
279
280 # Create bar chart (histogram)
281 fig = px.bar(
282 x=amino_acids,
283 y=counts,
284 labels={'x': 'Amino Acid', 'y': 'Frequency'},
285 title=f'Amino Acid Composition - PDB: {pdb_id.upper()}',
286 color=counts,
287 color_continuous_scale='Viridis'
288 )
289
290 # Update layout
291 fig.update_layout(
292 xaxis_title='Amino Acid (3-letter code)',
293 yaxis_title='Count',
294 showlegend=False,
295 height=400,
296 hovermode='x'
297 )
298
299 # Sort by amino acid name for consistent display
300 fig.update_xaxes(categoryorder='category ascending')
301
302 return fig
303
304# Run the app
305if __name__ == "__main__":
306 app.run(host='0.0.0.0', port=8050, debug=True)
Again, to run the updated app, simply execute the following command in your VS Code terminal (if it’s not already running):
(.venv) [mbs337-vm]$ python app.py
Dash is running on http://0.0.0.0:8050/
* Serving Flask app 'app'
* Debug mode: on
Now we can navigate to http://<IP_ADDRESS>:8050/ in our web browser to see the updated PDB dashboard
with the new reset button.
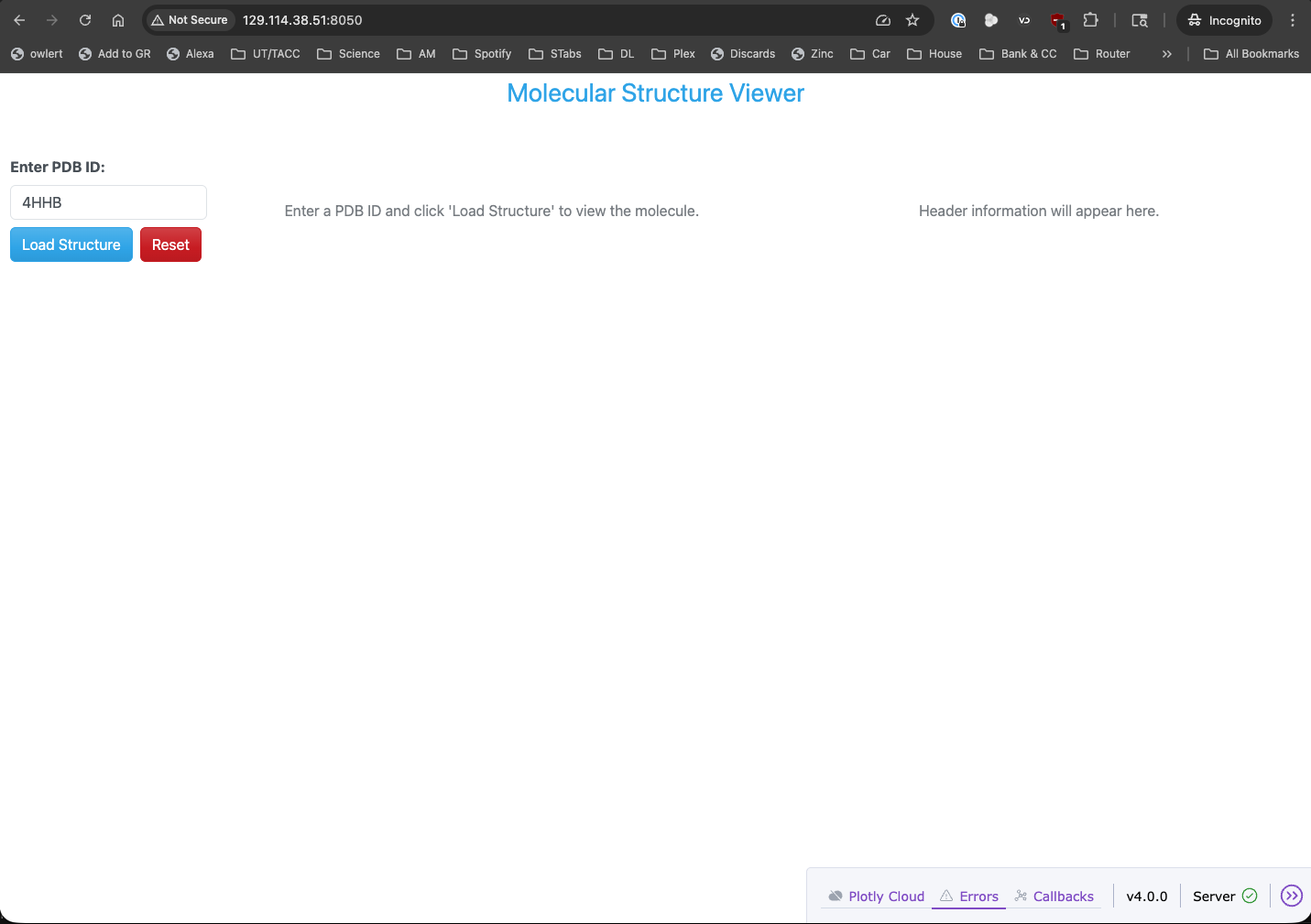
PDB dashboard application with reset button running in a web browser.