Working with Sequence Data (FASTQ)
In this hands-on module, we will learn how to work with the FASTQ data format. FASTQ extends FASTA by storing quality scores alongside sequence data from sequencing instruments.
After going through this module, students should be able to:
Identify and understand valid FASTQ files
Use Linux commands to safely examine FASTQ structure
Read FASTQ files in Python using Biopython and specify the correct quality encoding
Understand why quality encoding matters and when to use
SeqIO.parse
FASTQ Format Basics
FASTQ is an extension of the FASTA format that includes quality scores along with sequence data. It is typically used to store raw sequence reads from high-throughput sequencing technologies like Illumina platforms. FASTQ files additionally contain per-base quality scores indicating the confidence of each base call.
A FASTQ file ends in the .fq or .fastq file extension, and consists of one or
more sequence records. Each record contains exactly four lines:
Line 1: A sequence identifier starting with
@(at symbol)Line 2: The raw sequence data
Line 3: A separator line that starts with
+(plus symbol), optionally followed by the same identifier as line 1Line 4: Quality scores encoded as ASCII characters; must have same number of characters as line 2
Here is an example of a FASTQ file downloaded from the NCBI Sequence Read Archive (SRA). This example contains exactly one sequence record:
@SRR20340966.1 1 length=150
CNGCTGAGTTGCTCCTCCAAGGCCTCCAAACGAAGCTTCAAAGCGCTTCCATTCACCTCCTGCTCCTCCATGCGTCTGCCCAGCTCCACCGTCTGCGCACAGGCAGCTTCCACAGCAGGTGGAATTGCAGGTTCGACCACAGGAGGCGCT
+SRR20340966.1 1 length=150
F#FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFF:,:FFFFF:FFFFFFFF::FFFFFF:FFFFF,FFF,:FFFFFFFFFFFFF
In this example:
Line 1 starts with
@and contains the read identifier in NCBI’s FASTQ formatLine 2 contains the sequence itself (nucleotides: A, T, G, C, N for unknown/ambiguous)
Line 3 starts with
+and optionally repeats the identifier (often just+)Line 4 contains quality scores as ASCII characters (one character per base)
Note on FASTQ Quality Score Encoding:
Quality scores in FASTQ files are encoded using ASCII characters, but different sequencing platforms and software versions use different encoding schemes. This is crucial to understand when working with FASTQ files from different sources.
Quality scores represent the probability that a base call is incorrect:
Phred Quality Score (Q) |
Probability of incorrect base call |
Base call accuracy |
|---|---|---|
10 |
1 in 10 |
90% |
20 |
1 in 100 |
99% |
30 |
1 in 1000 |
99.9% |
40 |
1 in 10,000 |
99.99% |
50 |
1 in 100,000 |
99.999% |
60 |
1 in 1,000,000 |
99.9999% |
Different Platforms Use Different ASCII Encodings:
The quality score is then converted to a printable ASCII character by adding an offset. Different platforms use different offsets:
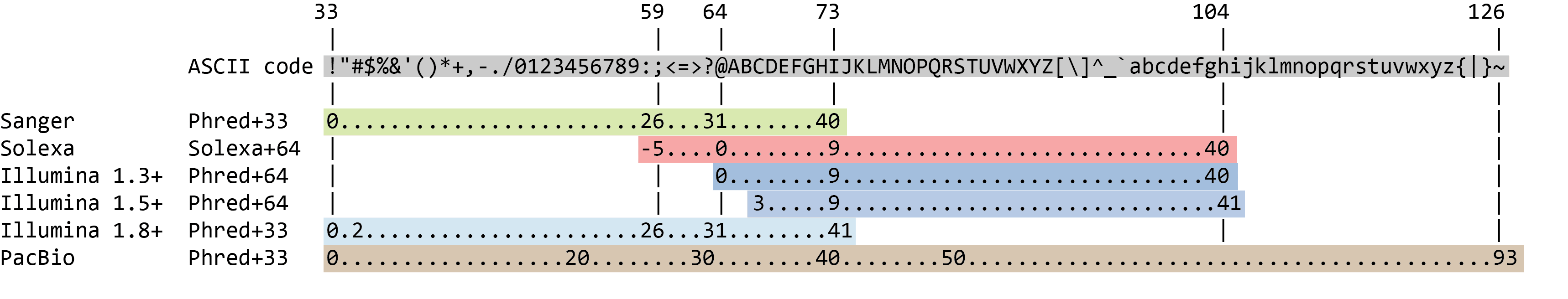
Different sequencers use different Phred quality score encodings.
While you may come across older datasets (Solexa/Illumina 1.3-1.7) that use Phred+64 offset, the current standard is Phred+33 (Sanger/Illumina 1.8+).
EXERCISE
See if you can answer the following questions about our example FASTQ sequence record:
@SRR20340966.1 1 length=150
CNGCTGAGTTGCTCCTCCAAGGCCTCCAAACGAAGCTTCAAAGCGCTTCCATTCACCTCCTGCTCCTCCATGCGTCTGCCCAGCTCCACCGTCTGCGCACAGGCAGCTTCCACAGCAGGTGGAATTGCAGGTTCGACCACAGGAGGCGCT
+SRR20340966.1 1 length=150
F#FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFF:,:FFFFF:FFFFFFFF::FFFFFF:FFFFF,FFF,:FFFFFFFFFFFFF
Can you tell which ASCII encoding scheme the above FASTQ file is using?
Which ASCII character corresponds to the worst Phred score for this encoding?
What is the Phred quality score of the majority of the base calls above?
What is the Phred quality score of the second base call?
Do you notice anything interesting about that second base call?
Attention
Many bioinformatics tools can auto-detect the encoding, but it is still essential for you to know your data source. Choosing the wrong encoding can lead to bad quality control and disastrous consequences down the line!
Working with FASTQ files
Let’s get some practice working with FASTQ files. We’ll use VSCode for these exercises.
Open a VSCode RemoteSSH session (Cmd+Shift+P -> RemoteSSH) and create a new terminal
(Cmd+Shift+P -> Terminal: Create New Terminal).
We’ll create an example FASTQ file within our working-with-bio-data directory.
Copy and paste the contents of this file into a new file called raw_reads.fastq
Helpful Linux Commands
Now let’s examine our FASTQ file. Your gut instinct may be to do a similar grep command that we used with the FASTA file
on the sequence identifier: grep "@" raw_reads.fastq. The problem with this is that @ could appear in line 4
since this line contains the ASCII-encoded quality scores, and @ is a valid ASCII symbol.
We need a method that understands the structure of the file, not just the characters in it.
Introducing awk
awk is a powerful text-processing command that works on one line at a time.
Unlike grep, which simply searches for matching text, awk allows us to perform actions
on specific lines within our file. The basic form of an awk command looks like this:
awk '{ action }' filename
where awk automatically reads the file line by line, and then applies the { action } to every line
(unless told otherwise).
It also provides a built-in variable called NR, which stands for Number of Records.
By default, one record = one line, and NR can therefore be used to represent the line number, starting at 1.
Since FASTQ files have a strict structure in which every read uses exactly 4 lines, that means that header lines always appear on lines 1, 5, 9, 13, etc. We can express this pattern mathematically:
NR % 4 == 1
Let’s test it out with an awk command:
awk 'NR % 4 == 1' raw_reads.fastq
This command will print every fourth line, starting with the first line, and thus prints only the FASTQ header lines and nothing else. Now we need a way to count these header lines to determine how many reads are in the file.
Introducing | and wc
In Linux, the pipe symbol (|) is used to connect two commands together. It will redirect the
output of the command on its left as the input to command on its right. It allows chaining
multiple commands together in a single line.
In addition, the wc command (short for word count) can be used to count the number of
words (or lines when we add the -l option) in a file from standard input:
To demonstrate this, we can combine awk and wc -l using a pipe:
awk 'NR % 4 == 1' raw_reads.fastq | wc -l
The number printed is the total number of read identifiers (and therefore sequencing reads) in the FASTQ file.
These command line tricks can be very useful to get a quick glance of the data we’re working with. For more advanced operations on our FASTQ files, we use Python and Biopython.
Read FASTQ from File
We can read FASTQ files in Python using SeqIO.parse from Biopython. Unlike the lightweight
SimpleFastaParser we used for FASTA, SeqIO.parse returns full SeqRecord objects that
include the sequence and its quality scores. This is appropriate when you need both sequence and
quality information (e.g., for quality control or filtering).
Attention
Memory and large files: SeqIO.parse is an iterator, so it does not load the entire
file into memory at once. However, each record is a full SeqRecord with sequence and quality
data. For very large FASTQ files (e.g., billions of reads), consider whether you need all
records in memory at once—if not, iterate and process in chunks; if you do (e.g., sorting),
be aware of memory limits.
You must tell Biopython which FASTQ quality encoding variant your file uses. If you use the wrong format, quality values will be wrong. Common format strings:
fastq-sanger— Phred+33 (Sanger, Illumina 1.8+). Use this for most modern Illumina data.fastq-illumina— Phred+64 (older Illumina 1.3–1.7).fastq-solexa— Solexa/Illumina legacy encoding.
We can actually use the information contained in the read identifier to help us identify the quality encoding variant:
@SRR20340966.1 1 length=150
The string SRRxxxxxxxx corresponds to a SRA Run Accession. We can copy that
run accession here to get more information about our data.
Our sample raw_reads.fastq was sequenced on the Illumina NovaSeq 6000 and was published in 2023.
It is very likely that this data uses the modern Phred+33 (Illumina 1.8+) encoding. We also determined
above that the existence of the # character confirms we are working with Phred+33 encoding.
Therefore, we will use fastq-sanger.
Activate your Python virtual environment and create a file called fastq_ex.py:
from Bio import SeqIO
with open('raw_reads.fastq', 'r') as f:
for record in SeqIO.parse(f, 'fastq-sanger'):
print(f"ID: {record.id}")
print(f"Sequence: {record.seq}")
print(f"Annot: {record.letter_annotations}")
break
ID: SRR20340966.1
Sequence: CNGCTGAGTTGCTCCTCCAAGGCCTCCAAACGAAGCTTCAAAGCGCTTCCATTCACCTCCTGCTCCTCCATGCGTCTGCCCAGCTCCACCGTCTGCGCACAGGCAGCTTCCACAGCAGGTGGAATTGCAGGTTCGACCACAGGAGGCGCT
Annot: {'phred_quality': [37, 2, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 11, 37, 37, 37, 25, 11, 25, 37, 37, 37, 37, 37, 25, 37, 37, 37, 37, 37, 37, 37, 37, 25, 25, 37, 37, 37, 37, 37, 37, 25, 37, 37, 37, 37, 37, 11, 37, 37, 37, 11, 25, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37]}
Each record has .id, .seq, and .letter_annotations (e.g., record.letter_annotations['phred_quality']
for a list of integer Phred scores per base).
Let’s see what would happen if we tried to use fastq-illumina instead of fastq-sanger:
from Bio import SeqIO
with open('raw_reads.fastq', 'r') as f:
for record in SeqIO.parse(f, 'fastq-illumina'):
print(f"ID: {record.id}")
print(f"Sequence: {record.seq}")
print(f"Length: {len(record.seq)}")
print(f"Annot: {record.letter_annotations}")
break
Traceback (most recent call last):
File "/home/ubuntu/mbs-337/working-with-bio-data/fastq_ex.py", line 4, in <module>
for record in SeqIO.parse(f, 'fastq-illumina'):
File "/home/ubuntu/mbs-337/myenv/lib/python3.12/site-packages/Bio/SeqIO/QualityIO.py", line 1113, in __next__
raise InvalidCharError(quality_string, invalid_index, details)
Bio.SeqIO.QualityIO.InvalidCharError: Invalid character (#) or (0x23) in quality string not in correct range (are you sure you're using the right QualityIO parser?) with context: [F#FFF...]
The output gives us an error because SeqIO.parse was expecting Phred+64 ASCII codes, and the # character is
not included in Phred+64.
EXERCISE
Let’s say this FASTQ file contains some RNA-Seq data we just got back from the sequencer. The first thing we need to do is check the quality of our data, and record it in a human-readable format like JSON. We’ll read in the FASTQ, define two Pydantic models (see Unit 2): one for each read summary (sequence identifier, sequence, total bases, average Phred quality), and one top-level model whose reads field holds a list of those summaries.
The structure we want looks like this:
{
"reads": [
{
"id": "SRR20340966.1 1 length=150",
"sequence": "CNGCTGAGTTGCTCCTCCAAGGCCTCCAAAC...",
"total_bases": 150,
"average_phred": 35.25
},
...
]
}
Let’s start by defining two Pydantic models: one for each read summary, and one (the top-level model) whose reads field will hold the list of read summaries:
import json
from Bio import SeqIO
from pydantic import BaseModel
# Define ReadSummary model
class ReadSummary(BaseModel):
id: str
sequence: str
total_bases: int
average_phred: float
# Define FastqSummary model (we'll create an instance of this with our list;
# .model_dump() will then convert that instance to a dict for JSON)
class FastqSummary(BaseModel):
reads: list[ReadSummary]
Now, let’s create an empty list to store our ReadSummary instances for each read.
We’ll use SeqIO.parse to extract the following from our FASTQ file:
The sequence identifier
The sequence itself
The length of the sequence
The average phred quality of the sequence
See if you can figure out this part before checking your work below:
Hint: Recall from the earlier example the format of record.letter_annotations:
{'phred_quality': [37, 2, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 11, 37, 37, 37, 25, 11, 25, 37, 37, 37, 37, 37, 25, 37, 37, 37, 37, 37, 37, 37, 37, 25, 25, 37, 37, 37, 37, 37, 37, 25, 37, 37, 37, 37, 37, 11, 37, 37, 37, 11, 25, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37, 37]}
# Create list of ReadSummary instances
reads_list = []
with open('raw_reads.fastq', 'r') as f:
for record in SeqIO.parse(f, 'fastq-sanger'):
# Calculate average phred quality
phred_scores = record.letter_annotations['phred_quality']
average_phred = sum(phred_scores) / len(phred_scores)
# Append information to reads_list
reads_list.append(ReadSummary(
id=record.id,
sequence=str(record.seq),
total_bases=len(record.seq),
average_phred=round(average_phred, 2)
))
We should end up with a list of ReadSummary instances that contain the information we need regarding each read:
print(reads_list[0])
id='SRR20340966.1' sequence='CNGCTGAGTTGCTCCTCCAAGGCCTCCAAACGAAGCTTCAAAGCGCTTCCATTCACCTCCTGCTCCTCCATGCGTCTGCCCAGCTCC
ACCGTCTGCGCACAGGCAGCTTCCACAGCAGGTGGAATTGCAGGTTCGACCACAGGAGGCGCT' total_bases=150 average_phred=35.51
Now we create a FastqSummary instance from our list (so we have one top-level object with a
reads field), convert that instance to a dictionary with .model_dump(), and write the
result to fastq_summary.json.
data = FastqSummary(reads=reads_list)
with open('fastq_summary.json', 'w') as outfile:
json.dump(data.model_dump(), outfile, indent=2)
print(f"Wrote {len(data.reads)} reads to fastq_summary.json")
Your new JSON file should look like this:
{
"reads": [
{
"id": "SRR20340966.1",
"sequence": "CNGCTGAGTTGCTCCTCCAAGGCCTCCAAACGAAGCTTCAAAGCGCTTCCATTCACCTCCTGCTCCTCCATGCGTCTGCCCAGCTCCACCGTCTGCGCACAGGCAGCTTCCACAGCAGGTGGAATTGCAGGTTCGACCACAGGAGGCGCT",
"total_bases": 150,
"average_phred": 35.51
},
{
"id": "SRR20340966.2",
"sequence": "CAGGATTCCCCAGCCTAGCGCCGGCTGCGCGGTGGGCCTTCCACCAAAGCCTTCCTCCAAGCTCCATACTGAATGTCAAACTCCGATTTCGTCAACTCTGAGCGAGGGGTCGCAGGATCTTCTGGTCTCCTGCCGGTCTCACGCCAATAC",
"total_bases": 150,
"average_phred": 36.75
}
]
}
Congratulations! You’ve just performed a real-world bioinformatics task and successfully saved your raw sequencing quality metrics to a new JSON file.
In this exercise, we:
Defined two Pydantic models:
ReadSummary (one per read)
FastqSummary (the top-level model with a
readsfield).
Built a list of ReadSummary instances (
reads_list)Created a FastqSummary instance with that list (
data = FastqSummary(reads=reads_list))Used
data.model_dump()to convert that instance to a plain Python dictionary andjson.dump()to write the JSON file.Pydantic validates the types (e.g.,
total_basesis an integer,average_phredis a float) automatically.
When you run the script, fastq_summary.json will contain one object per read with the
identifier, full sequence, total base count, and average Phred score. You can open the JSON
file to inspect the structure or use it as input for another script.